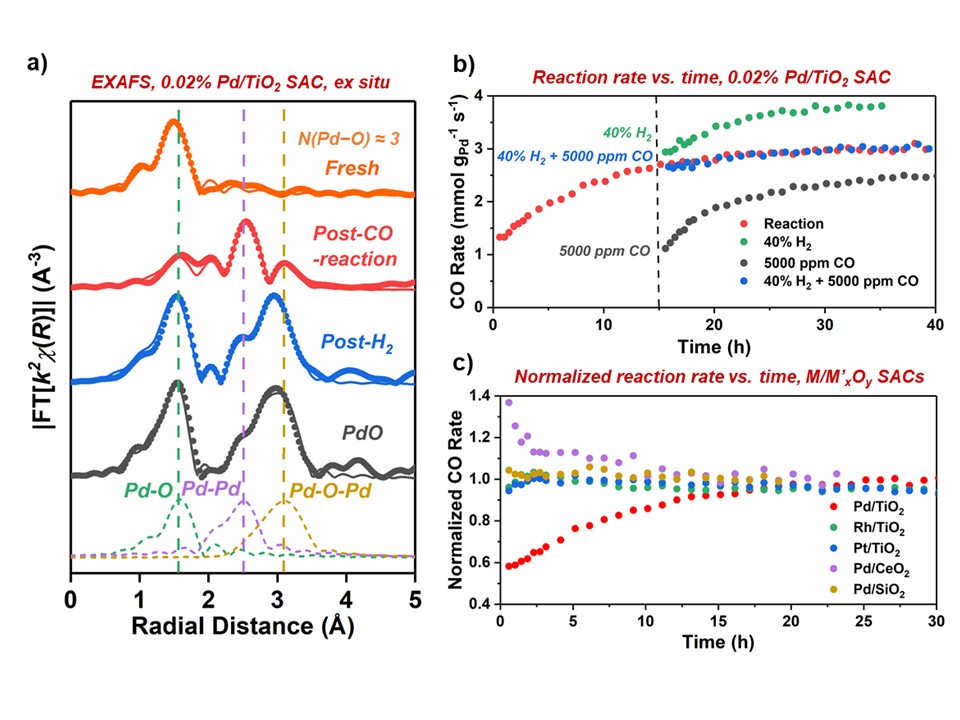
Interests in single-atom catalysts (SACs) have observed rapid increases recently. As a result of the instability and high mobility of single-atoms (SAs), the structure of SACs and the chemical environment of metal centers are often dynamic under reaction conditions, limiting their application scopes and challenging mechanistic investigations. Nonetheless, limited understanding has been obtained in the important area and systematic insights are desired. Here, we present detailed studies that elucidate the evolution of Pd SAs supported on anatase TiO
2 during high-temperature CO
2 hydrogenation, a highly enticing reaction for carbon mitigation and chemical production. We discovered that under the reaction conditions, low-surface-density Pd
1O
3 SAs (Figure a) on TiO
2 evolve in two pathways: forming a more active species, Pd
active, and sintering into large metallic particles. The former leads to the
in situ increase in the reaction rate over ~ 30 h (Figure b), has an activation barrier of ~121 kJ/mol, and is facilitated by H
2 in the reaction stream (Figure b). In contrast, the latter is induced by CO produced by the reaction, resulting in catalyst deactivation (Figure b). The
in situ Pd
active formation is unique to Pd/TiO
2, as other noble metal SAs (Pt, Rh, or Ru) and supports (CeO
2 or SiO
2) do not exhibit the same behavior (Figure c). The exact structure of Pd
active is under thorough investigation with
in situ spectroscopy and theory, and
ex situ XAS reveal that they can be completely oxidized into bulk-like PdO upon air exposure, while metallic particles created by CO treatment cannot (Figure a). This presentation depicts a complete and clear picture of the dynamic behaviors of TiO
2-supported Pd SAs under CO
2 hydrogenation and relevant reducing conditions. These insights will benefit both the understanding of hydrogenation mechanisms over SAs and the design of more stable, versatile SACs.